Shrinking Bat DNA and Elastic Genomes

Skip Sterling for Quanta Magazine
Introduction
Take an onion. Slice it very thin. Thinner than paper thin: single-cell thin. Then dip a slice in a succession of chemical baths cooked up to stain DNA. The dyed strands should appear in radiant magenta — the fingerprints of life’s instructions as vivid as rose petals on a marital bed. Now you can count how much DNA there is in each cell. It’s simply a matter of volume and density. A computer can flash the answer in seconds: 17 picograms. That’s about 16 billion base pairs — the molecular links of a DNA chain.
Maybe that number doesn’t mean much to you. Or maybe you’re scratching your head, recalling that your own hereditary blueprint weighs in at only 3 billion base pairs. “Huh?” joked Ilia Leitch, an evolutionary biologist at the Royal Botanic Gardens, Kew, in England. Her reaction mimicked the befuddlement of countless anthropocentric minds who have puzzled over this discrepancy since scientists began comparing species’ genomes more than 70 years ago. “Why would an onion have five times more DNA than we have? Are they five times more clever?”
Of course, it wasn’t just the onion that upended assumptions about a link between an organism’s complexity and the heft of its genetic code. In the first broad survey of animal genome sizes, published in 1951, Arthur Mirsky and Hans Ris —pioneers in molecular biology and electron microscopy, respectively — reported with disbelief that the snakelike salamander Amphiuma contains 70 times as much DNA as a chicken, “a far more highly developed animal.” The decades that followed brought more surprises: flying birds with smaller genomes than grasshoppers; primitive lungfish with bigger genomes than mammals; flowering plants with 50 times less DNA than humans, and flowering plants with 50 times more; single-celled protozoans with some of the largest known genomes of all.

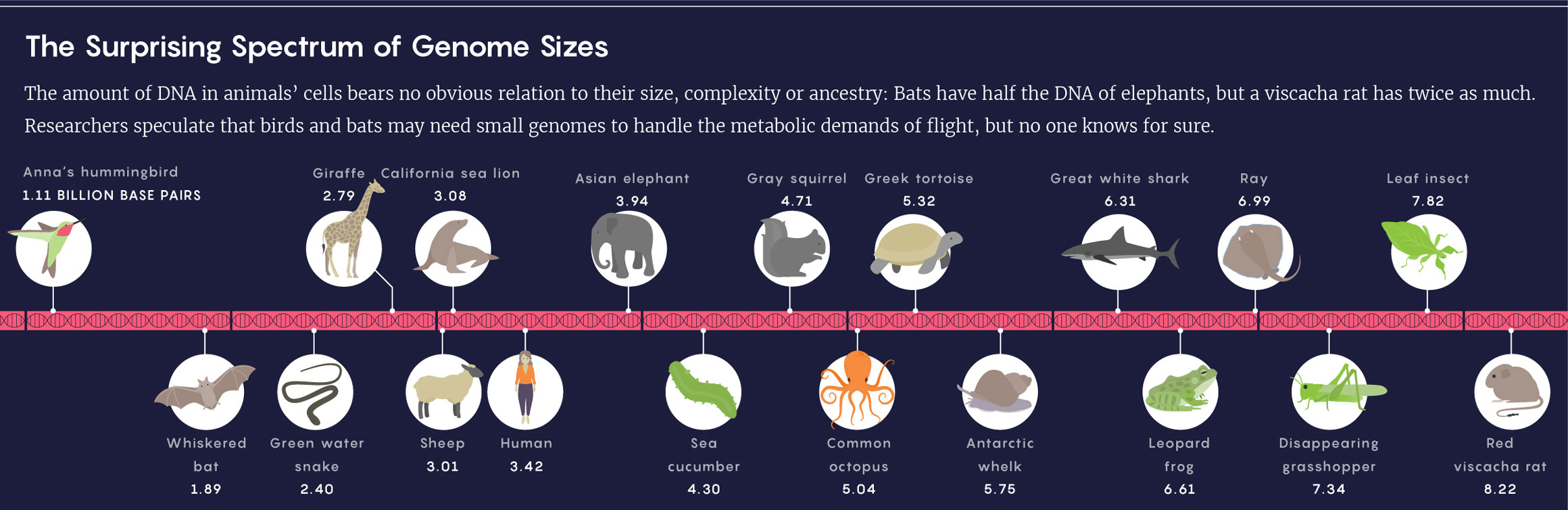
Lucy Reading-Ikkanda/Quanta Magazine; Source: Animal Genome Size Database
Even setting aside the genetic miniatures of viruses, cellular genome sizes measured to date vary more than a millionfold. Think pebbles versus Mount Everest. “It’s just crazy,” Leitch said. “Why would that be?”
By the 1980s, biologists had a partial answer: Most DNA does not consist of genes — those functional lines of code that translate into the molecules carrying out the business of a cell. “Large genomes have vast amounts of noncoding DNA,” Leitch said. “That’s what’s driving the difference.”
But although this explanation solved the paradox of the clever onion, it wasn’t particularly satisfying. “It just opened up more cans of worms,” said Ryan Gregory, a biologist at the University of Guelph who runs the online Animal Genome Size Database. Why, for instance, do some genomes contain very little noncoding DNA — also, controversially, often called “junk DNA” — while others hoard it? Does all this clutter — or lack of it — serve a purpose?
This past February, a tantalizing clue arose from research led by Aurélie Kapusta while she was a postdoctoral fellow working with Cedric Feschotte, a geneticist then at the University of Utah, along with Alexander Suh, an evolutionary biologist at Uppsala University in Sweden. The study, one of the first of its kind, compared genome sequences across diverse lineages of mammals and birds. It showed that as species evolved, they gained and shed astonishing amounts of DNA, although the average size of their genomes stayed relatively constant. “We see the genome is very dynamic, very elastic,” said Feschotte, who is now at Cornell University.
To explain this tremendous DNA turnover, Feschotte proposes an “accordion model” of evolution, whereby genomes expand and contract, forever gathering up new base pairs and dumping old ones. These molecular gymnastics represent more than a curiosity. They hint at hidden forces shaping the genome — and the organisms that genomes beget.
The Dynamics of DNA
The first signs that inheritance involves the transmission of more than just genes emerged around the time that Mirsky and Ris were marveling at the enormousness of the salamander genome. In the 1940s, a Swedish geneticist named Gunnar Östergren became fascinated with odd hereditary structures found in some plants. Östergren wrote that the structures, known as B chromosomes, appeared to have “no useful function at all to the species carrying them.” He concluded that these extraneous sequences were “genetic parasites” — hijackers of the “host” genome’s reproductive machinery. Three decades later, the evolutionary biologist Richard Dawkins solidified this idea in his popular 1976 book The Selfish Gene; the theory was quickly adapted to explain genome size.
By then, scientists had learned that B chromosomes are only a tiny fraction of the molecular parasites making genomes fat. The most prolific freeloaders are mobile strings of DNA called transposons, identified in 1944 by Barbara McClintock, the groundbreaking cytogeneticist who was honored with a Nobel Prize for that discovery. Transposons are popularly known as “jumping genes,” although they are rarely in fact true genes. They can get passed down from one generation to the next or transmitted between species, like viruses, and they come in several flavors. Some encode enzymes that snip a transposon out of its place in a genome and paste it elsewhere. Others copy themselves by manufacturing RNA templates or stealing enzymes from other transposons. (“You can get parasites within parasites,” Gregory said.)
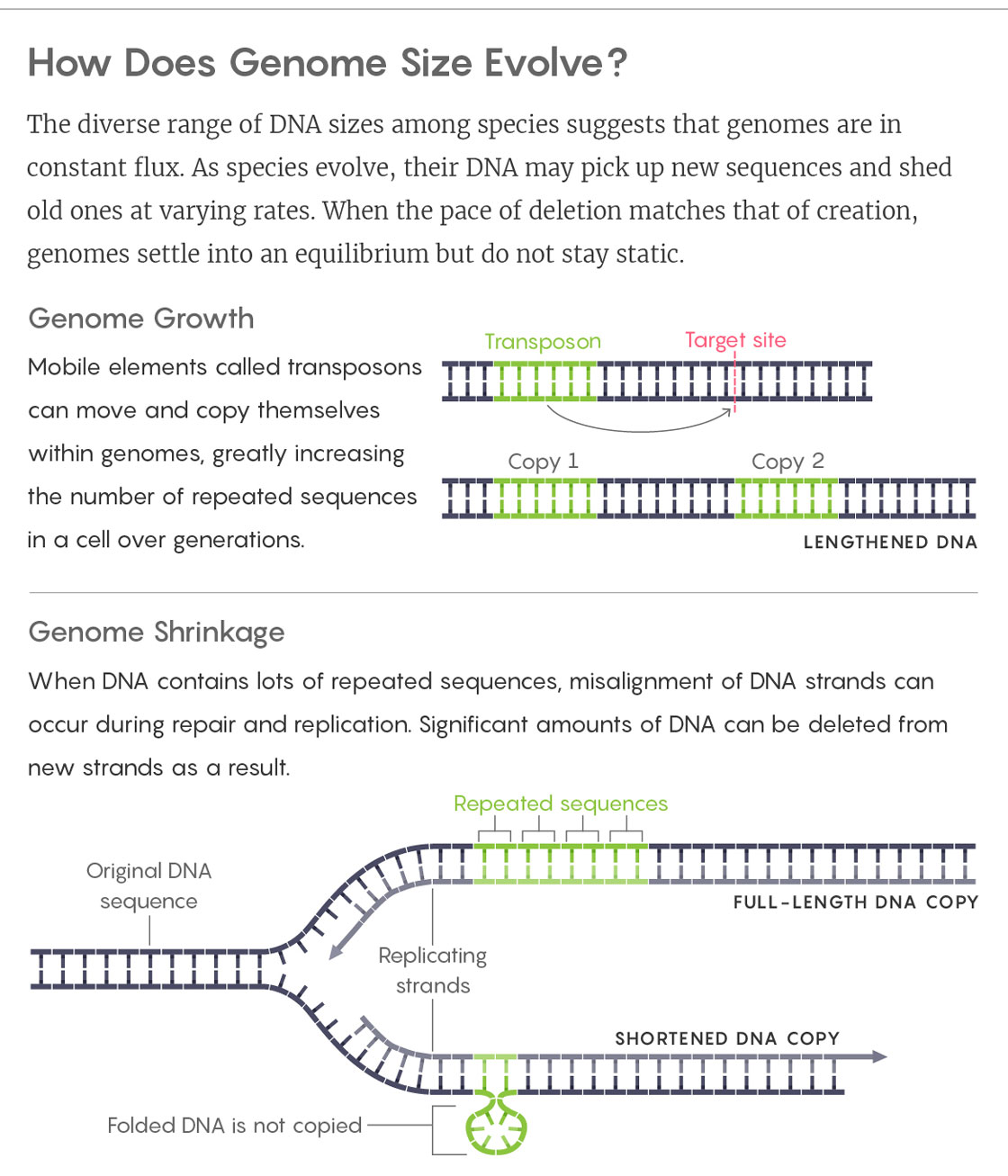
Lucy Reading-Ikkanda/Quanta Magazine
It’s not hard to see how these copies could quickly multiply, eventually taking over large portions of a genome. (More than 100 can pop up in a single generation of flies; they make up 85 percent of the maize genome and almost half of our own.) Proponents of the “selfish DNA” theory saw this pileup as the driving force of genome evolution: Within the ecosystem of a cell’s nucleus, natural selection would favor fast-multiplying transposons. But only up to a point. Once a genome reached a certain size, its bulk would start to interfere with an organism’s well-being — for example, by slowing the division of cells and thus the rate of the organism’s growth. Selection would kick in again, preventing further expansion. The limit would depend on the organism’s biology.
New evidence soon complicated this picture. In the late 1990s, Dmitri Petrov, then a doctoral student at Harvard University, began tracking small mutations in insects — random genetic changes of up to a few hundred base pairs that resulted from DNA damage, copying mistakes and poor strand repair. He started with flies. Analyzing defunct transposons, he showed that old code was being scrapped more quickly than new lines were being written (because random mutations are more likely to delete existing base pairs than to insert new ones). He wondered if this “deletion bias” might explain the fly’s relatively compact genome. He repeated the experiment in crickets and grasshoppers, whose genomes are, respectively, 10 and 100 times as large as the fly’s. This time, the deletion rates, although still dominant, were indeed considerably slower. Were some genomes bulkier than others simply because they weren’t as quick to clear out debris?
Based on these and similar observations, Petrov laid out a new model of genome size. Transposons, he argued, would always accumulate, sometimes very quickly. (Maize, for example, has doubled its genome in only 3 million years.) But over eons, small excisions would slowly chip away at a genome’s bulk. Eventually, the pace of expunction would match the pace of creation, and the genome would settle into equilibrium. Any number of forces in the chaotic nucleus might set — or reset — this balance.
Not everyone was convinced. Gregory, for one, maintained that spontaneous change happened too slowly to account for the dramatic morphing of genome size in many lineages. But no one could deny that loss was a powerful transformative force. As Gregory wrote in The Evolution of the Genome, “there are more complex interactions between [transposons] and their hosts than strict parasitism.” The tricky part was finding them.
The Fluttering Genomes of Bats
For Feschotte, the tip-off came from a bat. By the early 2000s, following advances in DNA sequencing, labs had begun decoding whole genomes and sharing the data online. At the time, Feschotte’s group was not particularly interested in the evolutionary dynamics of genome size, but they were extremely curious about what transposons could reveal about the history of life. So when the genome of the common little brown bat (Myotis lucifugus), the first genome sequence from a bat, appeared in 2006, Feschotte was thrilled. Bats have strikingly small genomes for a mammal — they’re more like those of birds — and it seemed likely they would hold surprises.

The tiny red viscacha rat has the largest known genome of any mammal.
Michael A. Mares

Meanwhile, southern bent-wing bats have some of the smallest mammalian genomes, despite resembling the viscacha rat in size and complexity.
Steve Bourne
The tiny red viscacha rat (left) has the largest known genome of any mammal. Meanwhile, southern bent-wing bats (right) have some of the smallest mammalian genomes, despite resembling the viscacha rat in size and complexity.
Michael A. Mares; Steve Bourne
Parsing the creature’s 2 billion base pairs, Feschotte and his colleagues did stumble on something strange. “We found some very weird transposons,” he said. Because these oddball parasite sequences didn’t appear in other mammals, they were likely to have invaded after bats diverged from other lineages, perhaps picked up from an insect snack some 30 to 40 million years ago. What’s more, they were incredibly active. “Probably 20 percent or more of the bat’s genome is derived from this fairly recent wave of transposons,” Feschotte said. “It raised a paradox because when we see an explosion of transposon activity, we’d predict an increase in size.” Instead, the bat genome had shrunk. “So we were puzzled.”
There was only one likely explanation: Bats must have jettisoned a lot of DNA. When Kapusta joined Feschotte’s lab in 2011, her first project was to find out how much. By comparing transposons in bats and nine other mammals, she could see which pieces many lineages shared. These, she determined, must have come from a common ancestor. “It’s really like looking at fossils,” she said. Researchers had previously assembled a rough reconstruction of the ancient mammalian genome as it might have existed 100 million years ago. At 2.8 billion base pairs, it was nearly human-size.
Next, Kapusta calculated how much ancestral DNA each lineage had lost and how much new material it had gained. As she and Feschotte suspected, the bat lineages had churned through base pairs, dumping more than 1 billion while accruing only another few hundred million. Yet it was the other mammals that made their jaws drop.
Mammals are not especially diverse when it comes to genome size. In many animal groups, such as insects and amphibians, genomes vary more than a hundredfold. By contrast, the largest genome in mammals (in the red viscacha rat) is only five times as big as the smallest (in the bent-wing bat). Many researchers took this to mean that mammalian genomes just don’t have much going on. As Susumu Ohno, the noted geneticist and expert in molecular evolution, put it in 1969: “In this respect, evolution of mammals is not very interesting.”
But Kapusta’s data revealed that mammalian genomes are far from monotonous, having reaped and purged vast quantities of DNA. Take the mouse. Its genome is roughly the same size it was 100 million years ago. And yet very little of the original remains. “This was a big surprise: In the end, only one-third of the mouse genome is the same,” said Kapusta, who is now a research associate in human genetics at the University of Utah and at the USTAR Center for Genetic Discovery. Applying the same analysis to 24 bird species, whose genomes are even less varied than those of mammals, she showed that they too have a lively genetic history.
“No one predicted this,” said J. Spencer Johnston, a professor of entomology at Texas A&M University. “Even those genomes that didn’t change size over a huge period of time — they didn’t just sit there. Somehow they decided what size they wanted to be, and despite mobile elements trying to bloat them, they didn’t bloat. So then the next obvious question is: Why the heck not?”
How DNA Gains Lead to Losses
Feschotte’s best guess points at transposons themselves. “They provide a very natural mechanism by which gain provides the template to facilitate loss,” he said. Here’s how: As transposons multiply, they create long strings of nearly identical code. Parts of the genome become like a book that repeats the same few words. If you rip out a page, you might glue it back in the wrong place because everything looks pretty much the same. You might even decide the book reads just fine as is and toss the page in the trash. This happens with DNA too. When it’s broken and rejoined, as routinely happens when DNA is damaged but also during the recombination of genes in sexual reproduction, large numbers of transposons make it easy for strands to misalign, and that slippage can result in deletions. “The whole array can collapse at once,” Feschotte said.
This hypothesis hasn’t been tested in animals, but there is evidence from other organisms. “It’s not so different from what we’re seeing in plants with small genomes,” Leitch said. “DNA in these species is often dominated by just one or two types of transposons that amplify and then get eliminated. The turnover is very dynamic: in 3 to 5 million years, half of any new repeats will be gone.”
That’s not the case for larger genomes. “What we see in big plant genomes — and also in salamanders and lungfish — is a much more heterogeneous set of repeats, none of which are present in [large numbers],” Leitch said. She thinks these genomes must have replaced the ability to knock out transposons with a novel and effective way of silencing them. “What they do is, they stick labels onto the DNA that signal to it to become very tightly condensed — sort of squished — so it can’t be read easily.” That alteration stops the repeats from copying themselves, but it also breaks the mechanism for eliminating them. So over time, Leitch explained, “any new repeats get stuck and then slowly diverge through normal mutation to produce a genome full of ancient degenerative repeats.”
Meanwhile, other forces may be at play. Large genomes, for instance, can be costly. “They’re energetically expensive, like running a big house,” Leitch said. They also take up more space, which requires a bigger nucleus, which requires a bigger cell, which can slow processes like metabolism and growth. It’s possible that in some populations, under some conditions, natural selection may constrain genome size. For example, female bow-winged grasshoppers, for mysterious reasons, prefer the songs of males with small genomes. Maize plants growing at higher latitudes likewise self-select for smaller genomes, seemingly so they can generate seed before winter sets in.
Some experts speculate that a similar process is going on in birds and bats, which may need small genomes to maintain the high metabolisms needed for flight. But proof is lacking. Did small genomes really give birds an advantage in taking to the skies? Or had the genomes of birds’ flightless dinosaur ancestors already begun to contract for some other reason, and did the physiological demands of flight then shrink the genomes of modern birds even more? “We can’t say what’s cause and effect,” Suh said.
It’s also possible that genome size is largely a result of chance. “My feeling is there’s one underlying mechanism that drives all this variability,” said Mike Lynch, a biologist at Indiana University. “And that’s random genetic drift.” It’s a principle of population genetics that drift — whereby a genetic variant becomes more or less common just by sheer luck — is stronger in small groups, where there’s less variation. So when populations decline, such as when new species diverge, the odds increase that lineages will drift toward larger genomes, even if organisms become slightly less fit. As populations grow, selection is more likely to quash this trait, causing genomes to slim.
None of these models, however, fully explain the great diversity of genome forms. “The way I think of it, you’ve got a bunch of different forces on different levels pushing in different directions,” Gregory said. Untangling them will require new kinds of experiments, which may soon be within reach. “We’re just at the cusp of being able to write genomes,” said Chris Organ, an evolutionary biologist at Montana State University. “We’ll be able to actually manipulate genome size in the lab and study its effects.” Those results may help to disentangle the features of genomes that are purely products of chance from those with functional significance.
Many experts would also like to see more analyses like Kapusta’s. (“Let’s do the same thing in insects!” Johnston said.) As more genomes come online, researchers can begin to compare larger numbers of lineages. “Four to five years from now, every mammal will be sequenced,” Lynch said, “and we’ll be able to see what’s happening on a finer scale.” Do genomes undergo rapid expansion followed by prolonged contraction as populations spread, as Lynch suspects? Or do changes happen smoothly, untouched by population dynamics, as Petrov’s and Feschotte’s models predict and recent work in flies supports?
Or perhaps genomes are unpredictable in the same way life is unpredictable — with exceptions to every rule. “Biological systems are like Rube Goldberg machines,” said Jeff Bennetzen, a plant geneticist at the University of Georgia. “If something works, it will be done, but it can be done in the most absurd, complicated, multistep way. This creates novelty. It also creates the potential for that novelty to change in a million different ways.”
This article was reprinted on Wired.com.



