The Cause of Depression Is Probably Not What You Think

Harol Bustos for Quanta Magazine
Introduction
People often think they know what causes chronic depression. Surveys indicate that more than 80% of the public blames a “chemical imbalance” in the brain. That idea is widespread in pop psychology and cited in research papers and medical textbooks. Listening to Prozac, a book that describes the life-changing value of treating depression with medications that aim to correct this imbalance, spent months on the New York Times bestseller list.
The unbalanced brain chemical in question is serotonin, an important neurotransmitter with fabled “feel-good” effects. Serotonin helps regulate systems in the brain that control everything from body temperature and sleep to sex drive and hunger. For decades, it has also been touted as the pharmaceutical MVP for fighting depression. Widely prescribed medications like Prozac (fluoxetine) are designed to treat chronic depression by raising serotonin levels.
Yet the causes of depression go far beyond serotonin deficiency. Clinical studies have repeatedly concluded that the role of serotonin in depression has been overstated. Indeed, the entire premise of the chemical-imbalance theory may be wrong, despite the relief that Prozac seems to bring to many patients.
A literature review that appeared in Molecular Psychiatry in July was the latest and perhaps loudest death knell for the serotonin hypothesis, at least in its simplest form. An international team of scientists led by Joanna Moncrieff of University College London screened 361 papers from six areas of research and carefully evaluated 17 of them. They found no convincing evidence that lower levels of serotonin caused or were even associated with depression. People with depression didn’t reliably seem to have less serotonin activity than people without the disorder. Experiments in which researchers artificially lowered the serotonin levels of volunteers didn’t consistently cause depression. Genetic studies also seemed to rule out any connection between genes affecting serotonin levels and depression, even when the researchers tried to consider stress as a possible cofactor.
“If you were still of the opinion that it was simply a chemical imbalance of serotonin, then yeah, it’s pretty damning,” said Taylor Braund, a clinical neuroscientist and postdoctoral research fellow at the Black Dog Institute in Australia who was not involved in the new study. (“The black dog” was Winston Churchill’s term for his own dark moods, which some historians speculate were depression.)
The realization that serotonin deficits by themselves probably don’t cause depression has left scientists wondering what does. The evidence suggests that there may not be a simple answer. In fact, it’s leading neuropsychiatric researchers to rethink what depression might be.
Treating the Wrong Disease
The focus on serotonin in depression began with a tuberculosis drug. In the 1950s, doctors started prescribing iproniazid, a compound developed to target lung-dwelling Mycobacterium tuberculosis bacteria. The drug wasn’t particularly good for treating tuberculosis infections — but it did bless some patients with an unexpected and pleasant side effect. “Their lung function and everything wasn’t getting much better, but their mood tended to improve,” said Gerard Sanacora, a clinical psychiatrist and the director of the depression research program at Yale University.
Perplexed by this outcome, researchers began studying how iproniazid and related drugs worked in the brains of rats and rabbits. They discovered that the drugs blocked the animals’ body from absorbing compounds called amines — which include serotonin, a chemical that carries messages between nerve cells in the brain.
Several prominent psychologists, among them the late clinicians Alec Coppen and Joseph Schildkraut, seized on the idea that depression could be caused by a chronic deficiency of serotonin in the brain. The serotonin hypothesis of depression went on to inform decades of drug development and neuroscientific research. During the late 1980s, it led to the introduction of selective serotonin reuptake inhibitor (SSRI) drugs, like Prozac. (The drugs raise levels of serotonin activity by slowing down the neurotransmitter’s absorption by neurons.) Today, the serotonin hypothesis is still the explanation most often given to patients with depression when they’re prescribed SSRIs.
But doubts about the serotonin model were circulating by the mid-1990s. Some researchers noticed that SSRIs often fell short of expectations and didn’t improve significantly on the performance of older drugs like lithium. “The studies didn’t really stack up,” Moncrieff said.


Merrill Sherman/Quanta Magazine
By the early 2000s, few experts believed that depression is caused solely by lack of serotonin, but no one ever attempted a comprehensive evaluation of the evidence. That eventually prompted Moncrieff to organize such a study, “so that we could get a view as to whether this theory was supported or not,” she said.
She and her colleagues found that it wasn’t, but the serotonin hypothesis still has adherents. Last October — just a few months after their review appeared — a paper published online in Biological Psychiatry claimed to offer a concrete validation of the serotonin theory. Other researchers remain skeptical, however, because the study looked at only 17 volunteers. Moncrieff dismissed the results as statistically insignificant.
A Different Chemical Imbalance
Although serotonin levels don’t seem to be the primary driver of depression, SSRIs show a modest improvement over placebos in clinical trials. But the mechanism behind that improvement remains elusive. “Just because aspirin relieves a headache, [it] doesn’t mean that aspirin deficits in the body are causing headaches,” said John Krystal, a neuropharmacologist and chair of the psychiatry department at Yale University. “Fully understanding how SSRIs produce clinical change is still a work in progress.”
Speculation about the source of that benefit has spawned alternative theories about the origins of depression.
Despite the “selective” in their name, some SSRIs change the relative concentrations of chemicals other than serotonin. Some clinical psychiatrists believe that one of the other compounds may be the true force inducing or relieving depression. For example, SSRIs increase the circulating levels of the amino acid tryptophan, a serotonin precursor which helps regulate sleep cycles. Over the last 15 years or so, this chemical has emerged as a strong candidate in its own right for staving off depression. “There’s quite good evidence from tryptophan depletion studies,” said Michael Browning, a clinical psychiatrist at the University of Oxford.
A number of tryptophan depletion studies found that about two-thirds of people who have recently recovered from a depressive episode will relapse when given diets artificially low in tryptophan. People with a family history of depression also appear vulnerable to tryptophan depletion. And tryptophan has a secondary effect of raising serotonin levels in the brain.
Recent evidence also suggests that both tryptophan and serotonin may contribute to the regulation of bacteria and other microbes growing in the gut, and chemical signals from these microbiota could affect mood. While the exact mechanisms linking the brain and gut are still poorly understood, the connection seems to influence how the brain develops. However, because most tryptophan depletion studies so far have been small, the matter is far from settled.
Other neurotransmitters like glutamate, which plays an essential role in memory formation, and GABA, which inhibits cells from sending messages to one another, may be involved in depression as well, according to Browning. It’s possible that SSRIs work by tweaking the amounts of these compounds in the brain.
Moncrieff sees the hunt for other chemical imbalances at the root of depression as akin to rebranding rather than a truly novel line of research. “I would suggest that they are still subscribing to something like the serotonin hypothesis,” she said — the idea that antidepressants work by reversing some chemical abnormality in the brain. She thinks instead that serotonin has such widespread effects in the brain that we may have trouble disentangling their direct antidepressant effect from other changes in our emotions or sensations that temporarily override feelings of anxiety and despair.
Genetic Answers
Not all theories of depression hinge on neurotransmitter deficiencies. Some look for culprits at the genetic level.
When the first roughly complete draft sequence of the human genome was announced in 2003, it was widely hailed as the foundation of a new era in medicine. In the two decades since then, researchers have identified genes that underlie a huge spectrum of disorders, including about 200 genes that have been linked to a risk of depression. (Several hundred more genes have been identified as possibly raising the risk.)
“It’s really important that people understand that there is a genetics of depression,” Krystal said. “Until very recently, only psychological and environmental factors were considered.”
Our knowledge of the genetics, however, is incomplete. Krystal noted that studies of twins suggest that genetics may account for 40% of the risk of depression. Yet the currently identified genes seem to explain only about 5%.
Moreover, simply having the genes for depression doesn’t necessarily guarantee that someone will become depressed. The genes also need to be activated in some way, by either internal or external conditions.
“There’s a false distinction that is sometimes drawn between environmental factors and genetic factors,” said Srijan Sen, a neuroscientist at the University of Michigan. “For most common traits of interest, both genetic and environmental factors play a critical role.”
Sen’s lab studies the genetic basis of depression by mapping subjects’ genomes and carefully observing how individuals with different genetic profiles respond to changes in their environment. (Recently, they have looked at stress brought on by the Covid-19 pandemic.) Different genetic variations can affect whether individuals respond to certain types of stress, such as sleep deprivation, physical or emotional abuse, and lack of social contact, by becoming depressed.
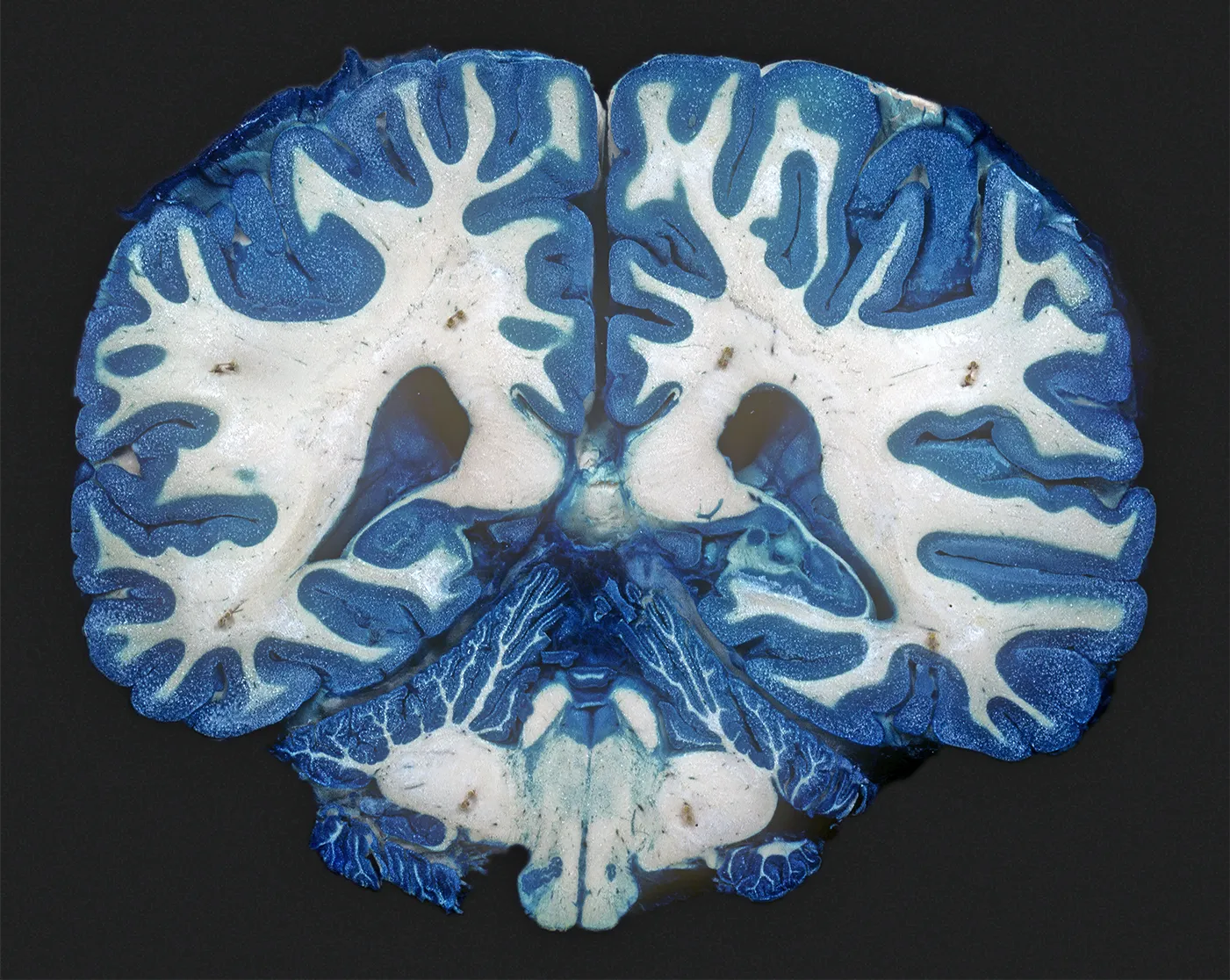
Research suggests that in the brains of people with chronic depression, the “white matter” areas that are rich in nerve fibers have fewer connections. The cause for this difference is uncertain, however.
Ralph T. Hutchins/Science Source
Environmental influences like stress can also sometimes give rise to “epigenetic” changes to a genome that affect subsequent gene expression. For example, Sen’s laboratory studies epigenetic changes in the caps on the ends of chromosomes, known as telomeres, which affect cell division. Other labs look at changes in chemical tags called methylation groups that can turn genes on or off. Epigenetic changes can sometimes even be passed down through generations. “The effects of the environment are just as biological as the effects of genes,” Sen said. “Just the source is different.”
Studies of these genes may someday help identify the form of treatment a patient would respond to best. Some genes may predispose an individual to better results from cognitive behavioral therapy, while other patients might fare better with an SSRI or therapeutic ketamine. However, it’s far too early to say which genes respond to which treatment, Sen said.
A Product of Neural Wiring
Differences in a person’s genes may predispose them to depression; so, too, may differences in the neural wiring and structure of their brain. Numerous studies have shown that individuals differ in how the neurons in their brains interconnect to form functional pathways, and that those pathways influence mental health.

Jonathan Repple and Susanne Meinert of Goethe University and their colleagues are exploring why chronically depressed people have fewer connections in their brains. Possible explanations include neuroplasticity and inflammation.
Roberto Schirdewahn; WWU/R
In a recent conference presentation, a team led by Jonathan Repple, a psychiatry researcher at Goethe University in Frankfurt, Germany, described how they scanned the brains of acutely depressed volunteers and found that they differed structurally from those of a non-depressed control group. For example, people experiencing depression showed fewer connections within the “white matter” of the nerve fibers in their brains. (However, there is no white-matter threshold for poor mental health: Repple notes that you can’t diagnose depression by scanning someone’s brain.)
After the depressed group underwent six weeks of treatment, Repple’s team ran another round of brain scans. This time, they found that the general level of neural connectivity in the depressed patients’ brains had increased as their symptoms lessened. To get the increase, it didn’t seem to matter what kind of treatment the patients received, so long as their mood improved.
A possible explanation for this change is the phenomenon of neuroplasticity. “Neuroplasticity means that the brain actually is able to create new connections, to change its wiring,” Repple said. If depression occurs when a brain has too few interconnections or loses some, then harnessing neuroplastic effects to increase interconnectedness might help lift a person’s mood.
Chronic Inflammation
Repple warns, however, that another explanation for the effects his team observed is also possible: Perhaps the depressed patients’ brain connections were impaired by inflammation. Chronic inflammation impedes the body’s ability to heal, and in neural tissue it can gradually degrade synaptic connections. The loss of such connections is thought to contribute to mood disorders.
Good evidence supports this theory. When psychiatrists have evaluated populations of patients who have chronic inflammatory diseases like lupus and rheumatoid arthritis, they’ve found that “all of them have higher-than-average rates of depression,” said Charles Nemeroff, a neuropsychiatrist at the University of Texas, Austin. Of course, knowing that they have an incurable, degenerative condition may contribute to a patient’s depressed feelings, but the researchers suspect that the inflammation itself is also a factor.
Medical researchers have found that inducing inflammation in certain patients can trigger depression. Interferon alpha, which is sometimes used to treat chronic hepatitis C and other conditions, causes a major inflammatory response throughout the body by flooding the immune system with proteins known as cytokines — molecules that facilitate reactions ranging from mild swelling to septic shock. The sudden influx of inflammatory cytokines leads to appetite loss, fatigue and a slowdown in mental and physical activity — all symptoms of major depression. Patients taking interferon often report feeling suddenly, sometimes severely, depressed.
If overlooked chronic inflammation is causing many people’s depression, researchers still need to determine the source of that inflammation. Autoimmune disorders, bacterial infections, high stress and certain viruses, including the virus that causes Covid-19, can all induce persistent inflammatory responses. Viral inflammation can extend directly to tissues in the brain. Devising an effective anti-inflammatory treatment for depression may depend on knowing which of these causes is at work.
It’s also unclear whether simply treating inflammation could be enough to alleviate depression. Clinicians are still trying to parse whether depression causes inflammation or inflammation leads to depression. “It’s a sort of chicken-and-egg phenomenon,” Nemeroff said.
The Umbrella Theory
Increasingly, some scientists are pushing to reframe “depression” as an umbrella term for a suite of related conditions, much as oncologists now think of “cancer” as referring to a legion of distinct but similar malignancies. And just as each cancer needs to be prevented or treated in ways relevant to its origin, treatments for depression may need to be tailored to the individual.
If there are different types of depression, they may present similar symptoms — such as fatigue, apathy, appetite changes, suicidal thoughts, and insomnia or oversleeping — but they might emerge from completely different mixes of environmental and biological factors. Chemical imbalances, genes, brain structure and inflammation could all play a role to varying degrees. “In five or 10 years, we won’t be talking about depression as a unitary thing,” Sen said.
To treat depression effectively, medical researchers may therefore need to develop a nuanced understanding of the ways it can arise. Nemeroff expects that someday the gold standard for care won’t be just one treatment — it will be a set of diagnostic tools that can determine the best therapeutic approach to an individual patient’s depression, be it cognitive behavioral therapy, lifestyle changes, neuromodulation, avoiding genetic triggers, talk therapy, medication or some combination thereof.
That prediction may frustrate some physicians and drug developers, since it’s much easier to prescribe a one-size-fits-all solution. But “appreciating the true, real complexity of depression takes us down a path that is ultimately going to be most impactful,” Krystal said. In the past, he said, clinical psychiatrists were like explorers who landed on a tiny unknown island, set up camp, and got comfortable. “And then we discovered that there’s this whole, enormous continent.”



