Quantum Complexity Tamed by Machine Learning
Introduction
In 2018, climate simulations were the third-largest use of computing cycles at a leading U.S. supercomputing cluster. The study of quarks and other subatomic particles came in second.
Topping the list was the most heavily cited idea in the physical sciences — though few have ever heard of it.
“It’s ridiculously important,” said Kieron Burke, a theoretical chemist at the University of California, Irvine. “It’s one of the most important things in science.”
Science’s best-kept secret goes by the name of density functional theory (DFT), and it is the chief method physicists and chemists use to understand just about anything more complicated than a hydrogen atom. For decades, researchers have harnessed DFT’s abilities to predict everything from the taste of coffee to the consistency of Jupiter’s core.
DFT gives scientists a powerful shortcut for predicting where electrons will go and, by extension, how atoms, molecules and other objects clothed in electrons will act. Physicists and chemists have long drawn on deep physical expertise to make their equations better reflect the intricate dance common to all electrons. But recently, new tools designed by neural networks are rivaling and, in some ways, outperforming their hand-crafted forerunners. Some researchers now believe machine learning could help researchers take larger and faster steps toward a master electron equation that would unlock the secrets of novel drugs, superconductivity and exotic materials.
“These machine learning methods,” said Marivi Fernández-Serra, a condensed matter physicist at Stony Brook University, “they got to where the field was in a couple of years and have already surpassed it.”
Electron Whisperers
To know electrons is to know the atoms, molecules and materials they make up. Physicists have thoroughly understood electrons since the 1920s, when Erwin Schrödinger published his eponymous equation. But when it comes to analyzing throngs of electrons, that understanding has proved all but useless.
The problem is that electrons are more clouds than particles. They spread out through space, overlapping with and jostling all other electrons in multiple ways. As their number rises, using Schrödinger’s wave equation to account for the constant contact between all electrons grows exponentially harder.
“Even for a few particles,” said Jeffrey Neaton, a condensed matter physicist at the University of California, Berkeley, “you just need more disk space than exists in the world right now even to write this wave function to a hard disk.”
In 1964, two physicists found a workaround. Pierre Hohenberg and Walter Kohn proved that they could perfectly capture every aspect of a molecule by smearing its electrons together into a contrived fluid — one that is thicker in some spots and thinner in others. The density of this electron juice contains all the information on the molecule’s complex wave function, they showed, freeing physicists from the impossible task of tracking electrons individually.
“That is the great miracle,” said Douglas Natelson, a professor of materials science at Rice University.
Hohenberg and Kohn’s work proved the existence of a powerful master equation — the universal density functional. The equation would take some instance of an electron soup and calculate its energy, guiding physicists to the most natural arrangement of electrons (the one with the lowest possible energy). The functional would be universal in the sense that it could, in principle, describe any system — from silicon bricks down to water molecules.
The only catch was that no one had any idea what the equation looked like.
Soon Kohn and another physicist, Lu Jeu Sham, made a first pass at writing down a practical density functional. An exact functional capturing all subtle aspects of electron behavior should be unspeakably complicated, so they split it in two. The first half was the part they knew how to do. It provided the energy of a group of electrons that could sense each other only on average. Everything else — the excess energy from the byzantine quantum effects and nonlocal interactions — went into the second half: a fudge factor known as the exchange and correlation functional.
They “reduced the thing that we don’t understand about the functional to one small part of it,” Neaton said.
Over the following decades, scientists built on Kohn and Sham’s work with increasingly clever ways of estimating the fudge factor, and density functionals became the de facto way to understand electrons. Researchers use them to predict how likely an atom is to grab or release an electron, the ways in which molecules vibrate (information the Curiosity rover uses to look for signs of life on Mars), the arrangement of atoms in crystal lattices, the speed of sound in a material, and much more. The theory’s endless stream of applications won Kohn a Nobel prize in 1998.
The Functional Ladder
As researchers ask for more precision from DFT, they’ve had to reckon with the ignorance stuffed into the exchange and correlation term of the functional, sharpening the details of their sketch to bring it more in line with the universal density functional.
Aron Cohen, a theoretical chemist at the artificial intelligence company DeepMind, is among those searching for the mythical universal equation. To stay grounded in the abstract landscape of theoretical quantum chemistry, he keeps a blue 3D-printed desk toy on hand. It looks like a plastic fortune cookie, and its curves trace out the exact shape of a functional that is universal, but only for the simplest of systems: It can be used to reveal anything one might wish to know about two electrons shared between any two atoms. Still, it reminds him that a functional that can handle any number of electrons and atoms may exist. “The thing we’re looking for is like this,” he said; it’s just much more complicated. “It’s a real thing.”
A major goal of DFT research is to find more accurate approximations of that universal functional. John Perdew, a physicist at Temple University and a leading functional developer, has long spearheaded this work. He describes the path toward the universal functional as like climbing the rungs on a ladder. On each rung, physicists add new ingredients to the functional. The simplest ingredient is just the thickness of the electron stew in each location. On the next rung, the functional also considers how quickly the thickness changes from place to place, giving researchers a broader view and making the functional more precise.
A key part of Perdew’s strategy is to use physical reasoning to identify certain mathematical properties that good approximations must obey, known as “exact constraints.” Higher rungs satisfy more of these constraints, and researchers have to search harder to find equations that obey them all.
Perdew’s group started tackling third-rung functionals, which blend six ingredients, in 1999, and in 2015, he released a state-of-the-art functional called SCAN. It was his eighth attempt, and the first to obey all 17 known constraints relevant on the third rung. Applicable to both molecules and solids, SCAN has proved one of the most capable approximations of the universal functional yet discovered.
“There are very rich possibilities for third-rung functionals,” Perdew said. “It takes time to figure out what is important and what works.”
Enter the Machines
As Perdew refined the art of improving density functionals with physical intuition, a revolution was brewing. Could algorithms ferret out patterns in electron behavior too subtle for humans to write down mathematically?
In 2012, Burke and his collaborators made the first modern attempt to apply machine learning to a set of simplified electrons. His one-dimensional prototype motivated him and other groups to see if algorithms could hoist researchers further up the ladder.
One breakthrough came in early 2021, when Burke and collaborators set up a neural network for a toy problem that could keep track of density errors as well as energy errors in a way that most previous attempts had ignored. “To get a functional which gives you both density and energy, you need a very flexible architecture,” said Alexandre Tkatchenko, a theoretical chemist at the University of Luxembourg. “It’s not easy to write a functional form with your mind.”
Fernández-Serra at Stony Brook used a similar strategy to design a neural network that would study a range of molecules and energies and search for a third-rung functional obeying most known constraints, essentially using a machine to retrace Perdew’s footsteps.
The resulting exchange and correlation functional beat SCAN in predicting the energies of unfamiliar molecules by roughly 10%, as she and Sebastian Dick reported in Physical Review B in the fall of 2021. But the modest gain suggested that Perdew’s work had already come close to finding the third rung’s ceiling.
“Physical intuition has somehow exhausted almost all that you can reach,” Fernández-Serra said. “Perdew’s functional really is as good as you can get without using machine learning.”
Climbing higher would require more complicated inputs — and algorithms to make sense of them.
DeepMind Scales the Ladder
Shortly before the pandemic, researchers at DeepMind, having conquered the board game Go, were searching for new challenges. They were especially interested in scientific machine learning problems, and Cohen pitched them DFT. He had spent years studying exact functionals of simple systems, like the one he keeps on his desk. But they were nowhere near complex enough to describe the real world.
“It felt like a long path to get to something that would be useful for chemistry,” he said.
One weakness at the heart of DFT ate away at Cohen in particular. Current density functionals often smear out electrons too much. The problem is especially glaring for lopsided systems, where electrons should gather mostly on one molecule. DFT tends to spread out the electron soup more evenly across both molecules. A related problem appears in chemical reactions, when DFT gives incorrect energies for particles merging and pulling apart, even for cases as simple as hydrogen atoms. “It’s this workhorse,” Cohen said, “but I know it has these massive failures.”
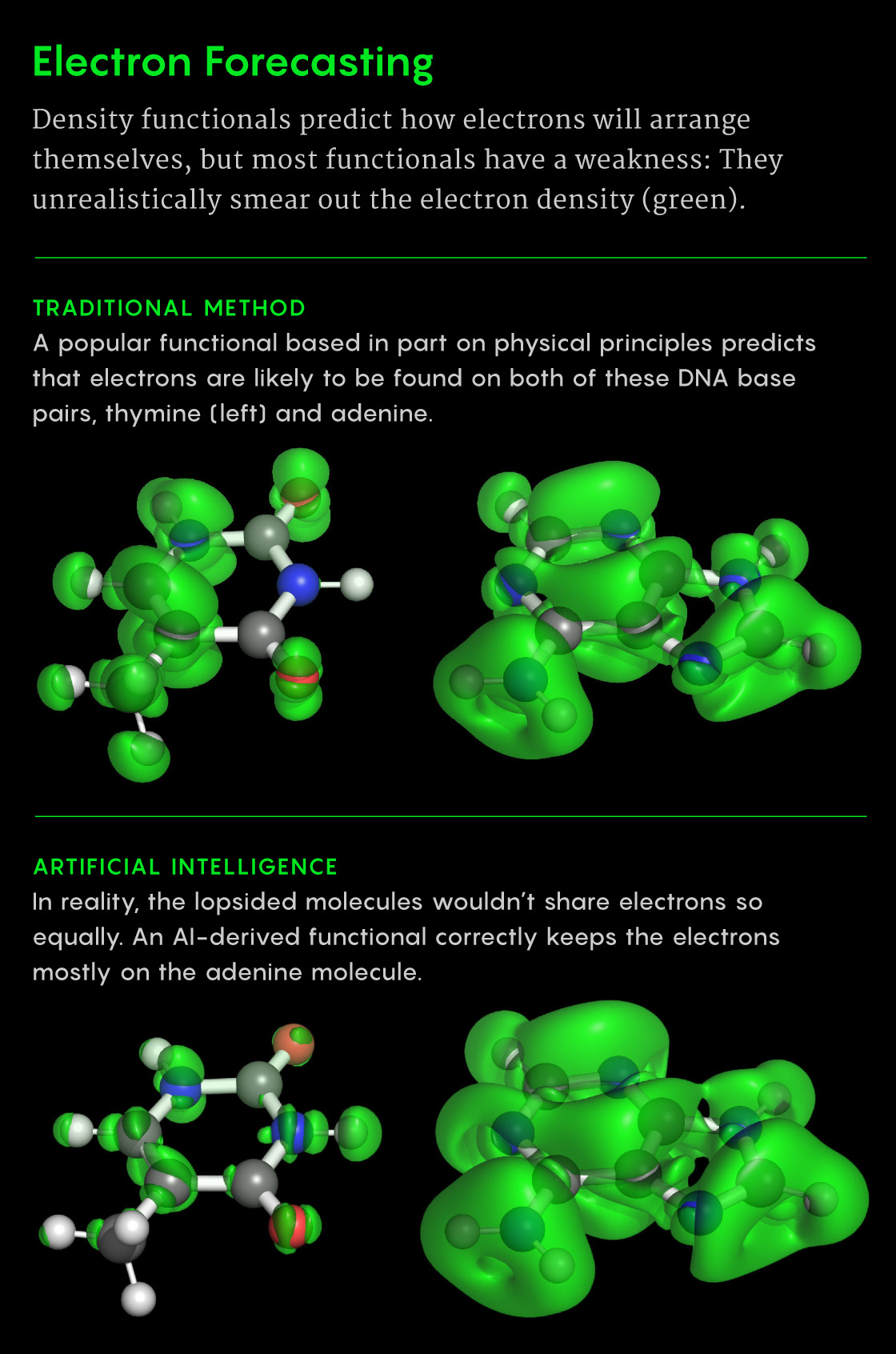
Samuel Velasco/Quanta Magazine; source: doi: 10.1126/science.abj6511
To design their next-generation functional, Cohen and the DeepMind team opted not to worry so much about satisfying a long list of physical principles. Instead, they’d lean on data, lots of data. They scoured the literature for databases of thousands of molecules with known energies (calculated at great cost using the Schrödinger equation or similar methods). Then they went further, using supercomputers to crunch the energies of hundreds of additional molecules — many of which took days to calculate.
While the team assembled an exhaustive library of example molecules, Cohen and other chemists decided how the functional should be structured.
They landed on a tantalizingly versatile approach. Over years of trial and error, researchers had found a special recipe for estimating part of the exchange and correlation functional. There was a sweet spot in performance when 80% of the energy for one piece of the functional was calculated in one way and 20% in another. Researchers had long suspected that the next step would be to let the 80/20 ratio vary from point to point around a molecule, but no one had fully succeeded in doing this.
There have been “100 papers maybe where people played with this form, but they hadn’t produced something that everybody could use,” said Burke, who introduced this type of functional in 1998. “Maybe it’s too hard for a person.”
With its sea of example molecules and the machine learning expertise of the DeepMind team, the group’s neural network was able to train a flexible fourth-rung functional of just that type. It can estimate the energies of a wide array of molecules better than SCAN and other top competitors, largely because it places electrons more accurately and better describes their spins. The functional, dubbed DM21, is the first general-purpose functional capable of handling the breaking and forming of chemical bonds. On December 9, the group described their functional in Science.
“This is the first reasonably general-purpose functional,” said Burke, who was not involved. “If this thing is as good as it looks, thousands of people will start using it within a year.”
However, Burke cautions that fully testing the functional will take time. The field is littered with the wreckage of functionals that initially looked promising but hid fatal flaws, and researchers have only just begun to pick apart DM21.
One weakness is that DM21 trained on molecules from the first three rows of the periodic table, where data is more plentiful. That means the electron behavior it learned may not carry over to metal atoms or solid materials, both of which are crucial for analyzing the copper-based family of high-temperature superconductors, for instance. The details of the universal functional representing those systems remain, for now, better approximated by SCAN and other functionals.
“For the foreseeable future, I don’t think there will be one functional that will do it all,” Tkatchenko said.
Toward a Universal Functional
The development of new functionals like those of Fernández-Serra and DeepMind suggests that machine learning can be a powerful tool for exploring new regions of the universal density functional, particularly those corresponding to molecules and chemistry.
It’s good for “really adjusting for the part of chemical space you want to address and making this functional as efficient as possible,” said Tkatchenko. “I think machine learning techniques really are here to stay.”
But whether improved chemistry functionals will reveal general features relevant to everything from atoms to materials remains to be seen. Perdew, for example, continues to search for new intuitive qualities that can further refine functionals in the traditional way. “I probably won’t put a lot of time into machine learning efforts,” he said, “because although machines can learn, they can’t yet explain to us what they have learned.”
Cohen hopes that DM21 has glimpsed universal features that could become enduring elements of future approximations, whether they’re generated by human minds or neural networks.
“The functional is infinitely complicated, so making any attack on it is good,” he said. “Ideally we’d like to unify them all.”



