Machine Intelligence Cracks Genetic Controls

Every cell in your body reads the same genome, the DNA-encoded instruction set that builds proteins. But your cells couldn’t be more different. Neurons send electrical messages, liver cells break down chemicals, muscle cells move the body. How do cells employ the same basic set of genetic instructions to carry out their own specialized tasks? The answer lies in a complex, multilayered system that controls how proteins are made.
Most genetic research to date has focused on just 1 percent of the genome — the areas that code for proteins. But new research, published today in Science, provides an initial map for the sections of the genome that orchestrate this protein-building process. “It’s one thing to have the book — the big question is how you read the book,” said Brendan Frey, a computational biologist at the University of Toronto who led the new research.
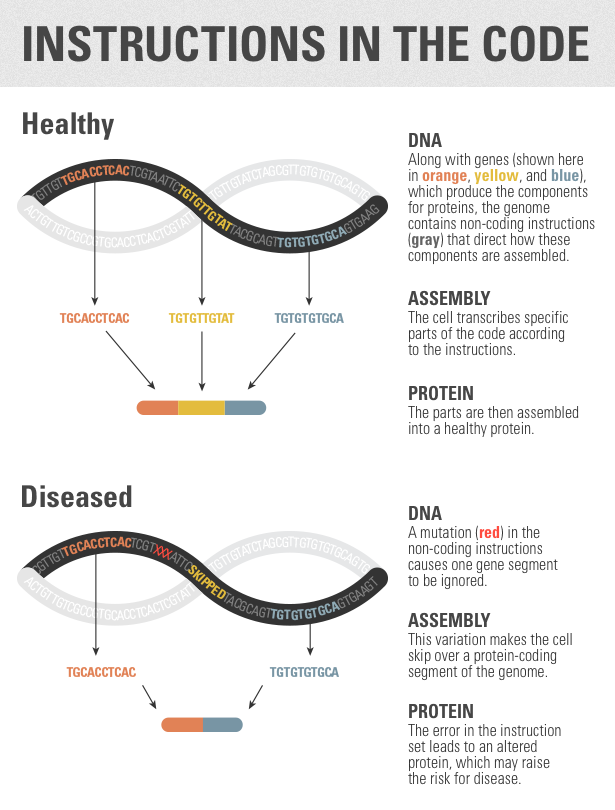
Frey compares the genome to a recipe that a baker might use. All recipes include a list of ingredients — flour, eggs and butter, say — along with instructions for what to do with those ingredients. Inside a cell, the ingredients are the parts of the genome that code for proteins; surrounding them are the genome’s instructions for how to combine those ingredients.
Just as flour, eggs and butter can be transformed into hundreds of different baked goods, genetic components can be assembled into many different configurations. This process is called alternative splicing, and it’s how cells create such variety out of a single genetic code. Frey and his colleagues used a sophisticated form of machine learning to identify mutations in this instruction set and to predict what effects those mutations have.
But the real significance of the research may come from the new tools it provides for exploring vast sections of DNA that have been very difficult to interpret until now. Many human genetics studies have sequenced only the small part of the genome that produces proteins. “This makes an argument that the sequence of the whole genome is important too,” said Tom Cooper, a biologist at Baylor College of Medicine in Houston, Texas.
Reading the Recipe
The splicing code is just one part of the noncoding genome, the area that does not produce proteins. But it’s a very important one. Approximately 90 percent of genes undergo alternative splicing, and scientists estimate that variations in the splicing code make up anywhere between 10 and 50 percent of all disease-linked mutations. “When you have mutations in the regulatory code, things can go very wrong,” Frey said.
“People have historically focused on mutations in the protein-coding regions, to some degree because they have a much better handle on what these mutations do,” said Mark Gerstein, a bioinformatician at Yale University, who was not involved in the study. “As we gain a better understanding of [the DNA sequences] outside of the protein-coding regions, we’ll get a better sense of how important they are in terms of disease.”
Scientists have made some headway into understanding how the cell chooses a particular protein configuration, but much of the code that governs this process has remained an enigma. Frey’s team was able to decipher some of these regulatory regions in a paper published in 2010, identifying a rough code within the mouse genome that regulates splicing. Over the past four years, the quality of genetics data — particularly human data — has improved dramatically, and machine-learning techniques have become much more sophisticated, enabling Frey and his collaborators to predict how splicing is affected by specific mutations at many sites across the human genome. “Genome-wide data sets are finally able to enable predictions like this,” said Manolis Kellis, a computational biologist at MIT who was not involved in the study.

Frey’s team used an approach called deep learning. Like any kind of machine-learning technique, the model tries to find a relationship between two sets of data. In this case, Frey’s team connected the human reference genome with rich data sets cataloging the amounts of different protein components in different tissues. (Just as two different cake recipes vary in their ratios of flour and sugar, brain cells and liver cells vary in how much of each protein they produce.) In essence, the algorithms trained a computational model to read instructions embedded in the DNA.
While scientists already knew how to read some aspects of the splicing code, the new model is unique. It allows scientists to predict how a wide array of genetic components will interact. “This group took what we knew about splicing and put it into a computational framework where we can weight all [the variables],” Burge said.
For example, researchers can use the model to predict what will happen to a protein when there’s a mistake in part of the regulatory code. Mutations in splicing instructions have already been linked to diseases such as spinal muscular atrophy, a leading cause of infant death, and some forms of colorectal cancer. In the new study, researchers used the trained model to analyze genetic data from people afflicted with some of those diseases. The scientists identified some known mutations linked to these maladies, verifying that the model works. They picked out some new candidate mutations as well, most notably for autism.
One of the benefits of the model, Frey said, is that it wasn’t trained using disease data, so it should work on any disease or trait of interest. The researchers plan to make the system publicly available, which means that scientists will be able to apply it to many more diseases.
A Broader Context
The model also reveals that when it comes to the genome, “context is important, just like in English,” Frey said. “‘Cat’ means different things whether we’re talking about pets or construction equipment.” In the same way, how the cell interprets a set of splicing instructions depends on other nearby instructions. A string of DNA that means “make lots of component X” might mean “don’t make component X” when it’s sitting near a second set of instructions. “Whether a sequence has an effect depends on whether another sequence has an effect,” Frey said. “Without understanding that, it’s hard to predict how a pattern will affect splicing.”
In addition, the model could help scientists reconsider known mutations, Burge said. Researchers already knew that some splicing instructions are found within protein-coding regions. In these cases, the same genetic sequence might code for both an ingredient and an instruction for what to do with it. (Consider whipped cream — it’s an ingredient, but it’s also in some ways an instruction.) A mutation in this protein-coding region might be dismissed as unimportant if it appears to do little or nothing to alter the corresponding protein. But when interpreted using the splicing code, that mutation might be found to have a profound effect by interfering with the splicing instructions. Frey’s group found many examples of these errors across the genome.
Frey hopes the model will ultimately prove useful for personalized medicine. For example, doctors cannot yet determine whether healthy people with novel mutations are predisposed to diseases like cancer. With further validation, Frey’s model might help to answer this question. “We can analyze any mutation, even those that haven’t been identified yet,” Frey said. This allows researchers to predict whether a novel mutation is likely to be dangerous or harmless — in essence, performing a screening test. “I want to see it have a huge impact on medicine,” he said. “I want to translate this into practice.”
This article was reprinted on Wired.com.