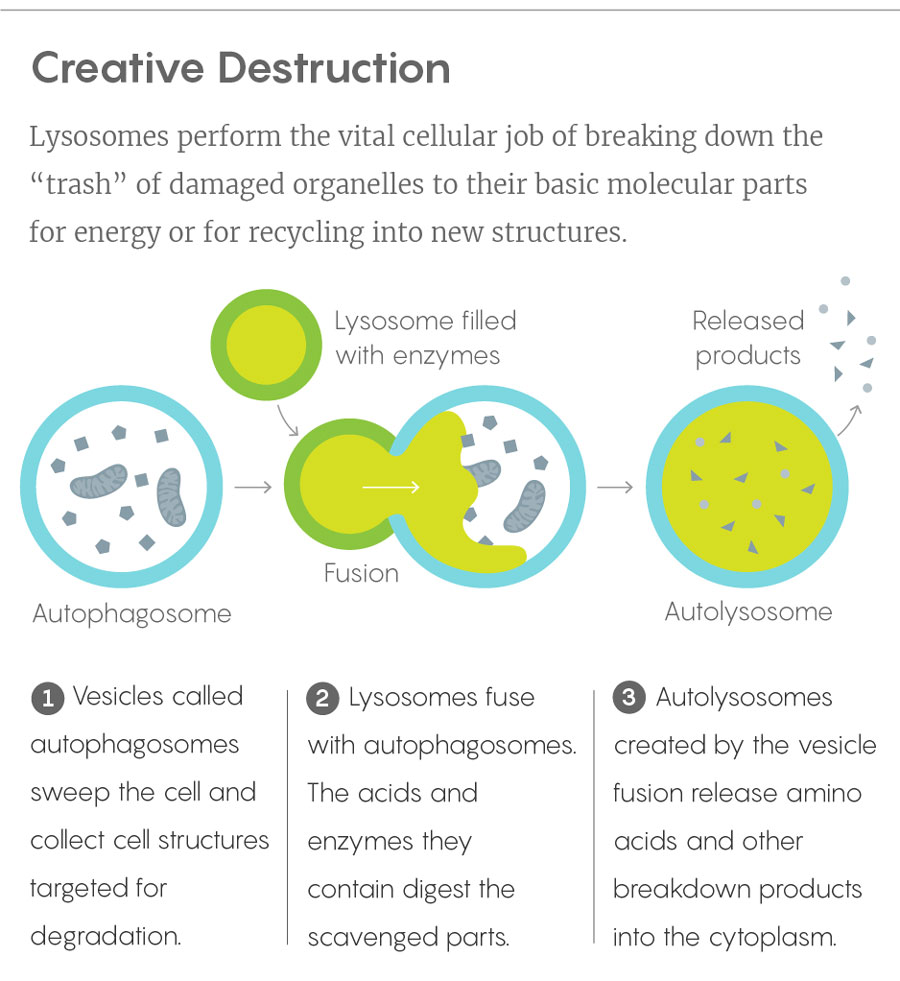
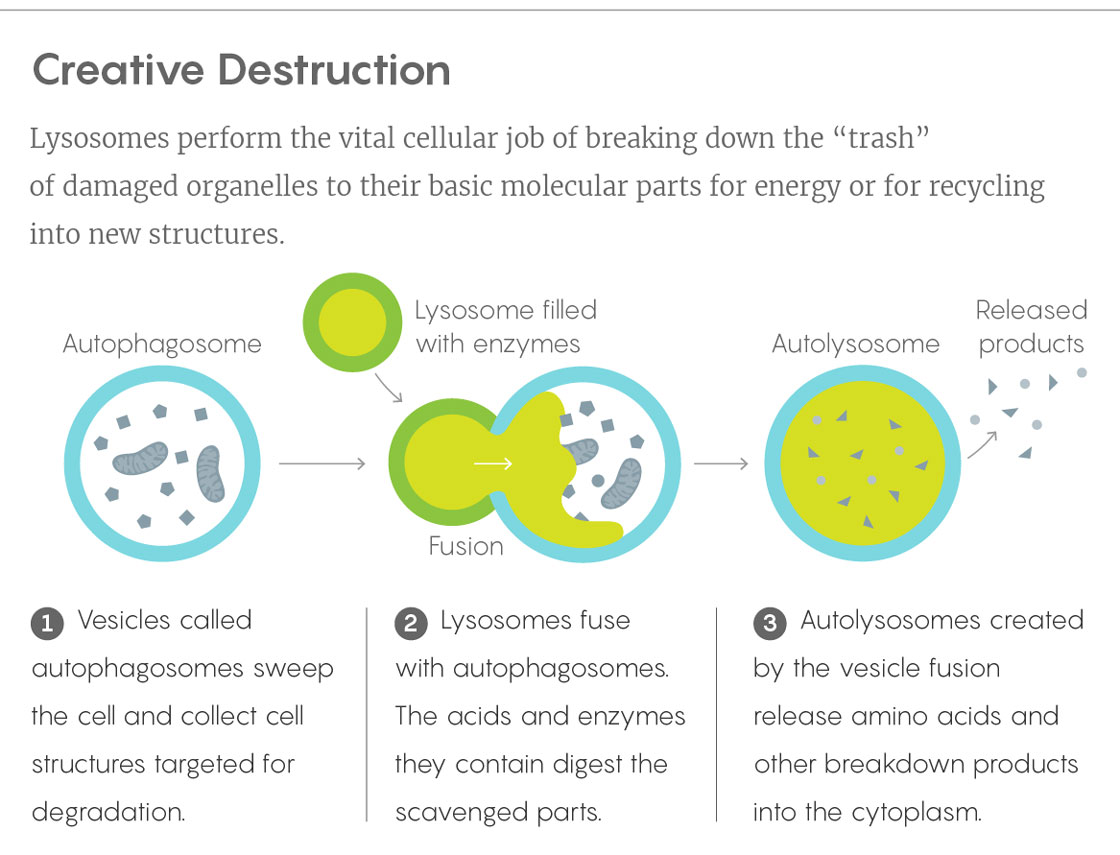
The Secret Power of the Cell’s Waste Bin
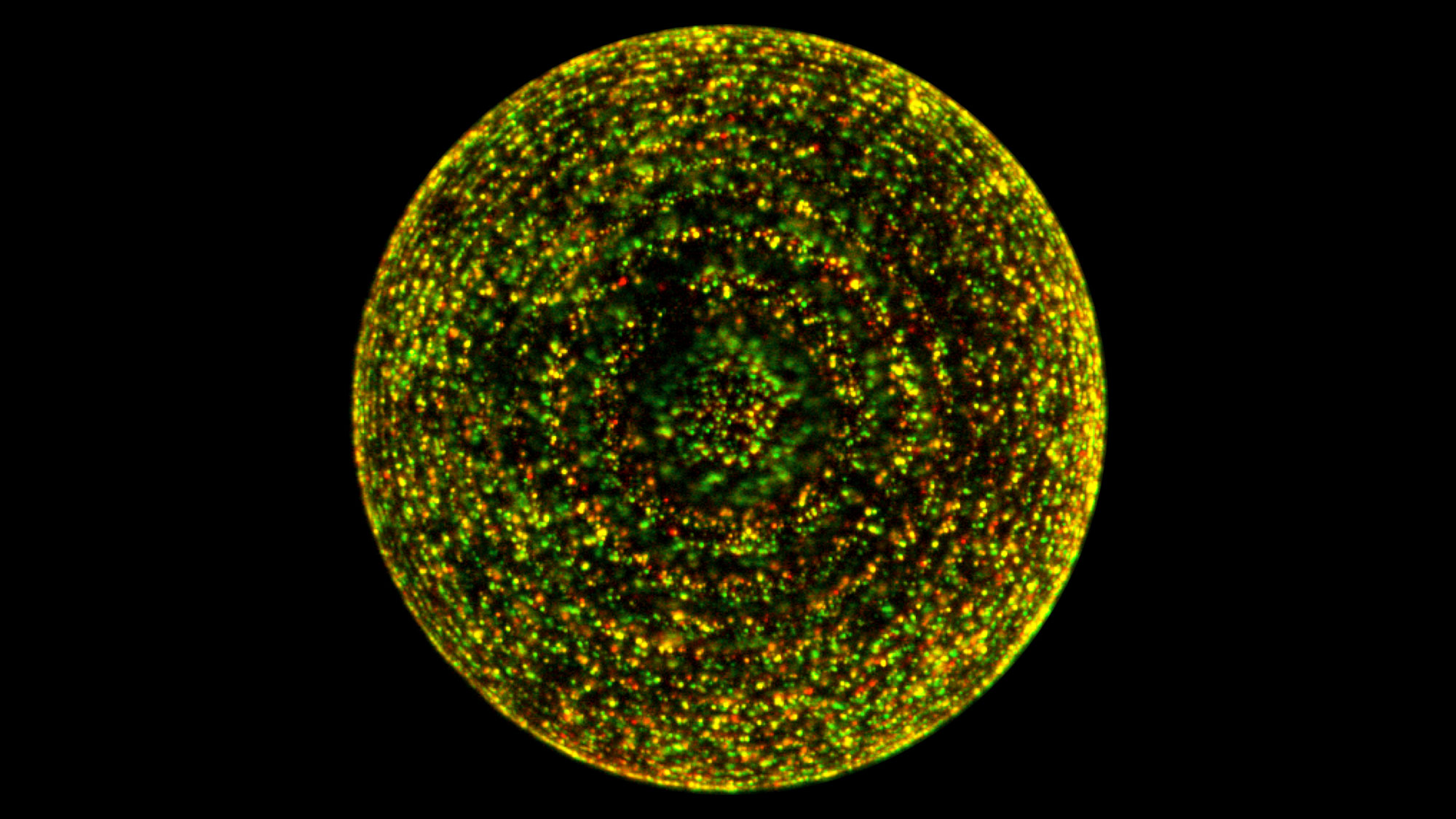
Glowing yellow dots mark where lysosomes (red) on beads in this preparation bind to the regulatory mTORC1 protein (green). But this binding occurs only when amino acids are present, which is the key to the regulatory influence of lysosomes.
Roberto Zoncu, Sabatini Lab, Whitehead Institute
Introduction
At a conference in Maine during the summer of 2008, the biochemist David Sabatini stood before an audience of his peers, prepared to dazzle them with a preview of unpublished results emerging from his lab at the Whitehead Institute for Biomedical Research in Cambridge, Massachusetts. The presentation did not go over well. His group was studying mTOR, a cellular enzyme he and colleagues had discovered more than a decade earlier. Among other things, they had tried to find out where mTOR aggregates inside cells, since this seemed likely to help explain the enzyme’s remarkable but mysterious influence over diverse cellular growth processes. Sabatini proudly projected a slide with the team’s findings, showing the enzyme arrayed along the surface of the organelles called lysosomes.
The audience was dubious. “People literally got up and said, ‘David, that’s the trash bin of the cell. It doesn’t make sense. Why decorate the outside of a trash can?” Sabatini recalled.
Over the nine years since Sabatini’s talk, lysosomes have won more respect. Research continues to show that lysosomes transcend the trash can role, acting as crucial advisers to the nucleus in its job of genetic regulation. That leap in status was obvious at the fourth Gordon Research Conference on Lysosomal Diseases, held March 5-10 in Barga, Italy. The lysosome was also celebrated in a paper that appeared last October in Annual Reviews of Cell and Developmental Biology, “The Lysosome as a Regulatory Hub.” Its authors, the San Francisco Bay Area researchers Rushika Perera and Roberto Zoncu, observed that recent studies have “raised the status of the lysosome from a catabolic dead end to a key signaling node, with far-reaching implications for our understanding of the logic of metabolic regulation both in health and in disease.”
Lucy Reading-Ikkanda/Quanta Magazine
In this loftier reckoning of lysosomes, the organelles deftly integrate metabolic information from throughout the cell and communicate it to the nucleus. Like snooping garbage collectors who learn the secrets of all the homeowners on their route, lysosomes gain a uniquely informed perspective on a cell’s status by picking through its molecular discards. And some of the finely tuned genetic controls of the nucleus would possibly be pilotless without them.
Lysosomes first drew attention in the 1950s, when the Belgian biochemist Christian de Duve stumbled across the saclike intracellular structures while trying to purify a protein found in rat livers. He named the previously unknown sacs after the Greek for “digestive body” because their contents were highly acidic and filled with enzymes that break down virtually any biomolecule that’s set before them. De Duve received a Nobel Prize for his discovery in 1974, but biologists were unenthusiastic about the organelle. Researchers nicknamed the lysosome “the recycle bin of the cell, or the trash can — nothing interesting,” said Zoncu, a biochemist at the University of California, Berkeley.
It wasn’t that lysosomes didn’t seem important — waste disposal systems inevitably are. They are responsible for digesting a cell’s damaged, malformed, superfluous or otherwise undesirable proteins and organelles, along with excess sugars and fats. When genetic defects cause lysosomes to make too little of any of the 60 or more enzymes associated with them, waste products pile up inside cells and cause lysosomal storage diseases, such as Tay-Sachs, Niemann-Pick and other disorders. Moreover, as a series of experiments led by Yoshinori Ohsumi (first at the University of Tokyo, then at Japan’s National Institute for Basic Biology) demonstrated in the 1990s, lysosomes are also instrumental in the vital process of autophagy, which allows cells to cannibalize their own organelles for resources in times of need and to combat the effects of illness and aging. That work brought Ohsumi a Nobel Prize in 2016 — a second Nobel to be awarded for work involving the cell’s lowly trash can.
But in the 1980s, when Andrea Ballabio, the founder of the Telethon Institute of Genetics and Medicine in Naples, was starting out in biological research, studies of the lysosome focused almost exclusively on what goes on inside it. He recalls the field as heavily and narrowly disease driven: Lysosome investigators purified enzymes that were deficient or dysfunctional in specific lysosomal storage disorders.

Andrea Ballabio, a geneticist at the University of Naples and scientific director of the Telethon Institute of Genetics and Medicine, studies how a transcription factor called TFEB increases production of key proteins in response to signals from the lysosome.
Photo courtesy of TIGEM
Ballabio had become interested in the lysosome while studying a particular kind of lysosomal storage disease. Multiple sulfatase deficiency causes scaly skin, stiff joints, seizures and developmental delays. The symptoms arise from mutations in a gene that, as Ballabio’s group discovered, is essential for the activation of a group of enzymes called sulfatases, many of which are lysosomal.
That discovery by Ballabio’s team, along with other studies of rare lysosomal disorders, convinced Ballabio that cells must have a system to boost lysosomal activity and a way to start making more lysosomes as cellular trash piles up. To do this, “you need to control the function of many different genes,” Ballabio said. He set out to find the master regulators that do this.
In 2009, his team reported that it had found an important one. They called it “transcription factor EB,” or TFEB. In the cell’s nucleus, TFEB binds to DNA sequences in many lysosomal genes and controls the rate at which they make proteins.
Precisely how TFEB’s activity could reflect the cell’s needs for lysosomes so comprehensively, however, was still unknown. But an answer would soon emerge from work that, at least initially, had nothing to do with lysosomes.
A Seat of Signaling
When Roberto Zoncu arrived as a postdoc at Sabatini’s lab at the Whitehead Institute in 2008, lysosomes were not uppermost in his mind. The lab’s focus was (and in many ways still is) on the enzyme that Sabatini had discovered in mammalian cells in 1994 and dubbed mechanistic target of rapamycin (mTOR). Implicated in aging and a slew of diseases including cancer and diabetes, mTOR signals cells to grow and divide under a surprisingly wide variety of circumstances. “One of the big motivating questions for us has been: How does that happen?” Sabatini said. “How does mTOR manage to sense so many things, integrate those signals and drive growth?”
A critical clue came when the team tracked the protein’s movements within cells. When cells were bathed in amino acid-free media, mTOR seemed to spread evenly throughout the cytoplasm. But if the media contained amino acids, within minutes mTOR moved into distinct clusters at specific locations inside the cell, shepherded there by other proteins called Rag GTPases. The enzymatic activity of mTOR depended on its reaching those locations, but the proteins that guided it there did not appear to turn it on. “We were stuck,” Sabatini said.
Zoncu therefore set out to learn what was special about where the mTOR protein was going in response to amino acids. In a key experiment, he stained cells with pairs of fluorescent antibodies: a red one that bound mTOR and a green one designed to bind to a protein associated with a different organelle in each round of the experiment. He then examined the cells under the microscope, looking for where the green and red fluorescent tags overlapped. This would indicate what else was located in the spots where the mTOR clustered.
Scanning the slide that stained for mitochondria — a potential target of huge metabolic importance — Zoncu found no overlap. He moved on to the slide for the next organelle, and the next. Still no overlap. “I almost lost hope,” Zoncu recalled.
Then came the lysosome slide. “All of a sudden, everything matched perfectly,” he said. The red mTOR staining and the green staining for lysosomal marker LAMP2 overlapped 100 percent.
Revving Up the Recycler
Those results added further support to the data Sabatini reported at the Maine conference in 2008 to his underwhelmed audience. But even Zoncu acknowledges that skepticism might have been warranted. Lysosomes, he says, could still “have just been a landing pad” — a convenient place for mTOR to touch down during activation.
Yet later experiments suggested otherwise. When Zoncu extracted lysosomes from cells and loaded them with amino acids, he saw that the more amino acids they carried, the more mTOR clustered on their surface and became active. (The enzyme mTOR forms two protein complexes in the cell; mTOR complex 1 [mTORC1] is the one found on lysosomes.) Those experiments, published in 2011, show that mTORC1 responds to the lysosomal contents, Sabatini says — as though the lysosomes tell mTORC1 about the amino acids they hold and mTORC1 adjusts its behavior accordingly.
Widening the Cellular Conversation
When Ballabio’s lab and Sabatini’s learned of one another’s results and joined forces, they soon worked out how the mTORC1 and TFEB pieces of the puzzle fit together, publishing the solution in 2012. In a healthy, well-fed cell, lysosomes have a cornucopia of proteins to break down to their amino acid components, and those amino acids work with proteins on the lysosome surface to anchor mTORC1 and activate it. The mTORC1 in turn keeps cytoplasmic TFEB out of the nucleus. When a cell becomes starved or stressed, mTORC1 drops away from the lysosome and TFEB is freed to bind its targets on the nuclear DNA. Acting as a master sensor of lysosomal function, TFEB turns on genes for more lysosomal enzymes.
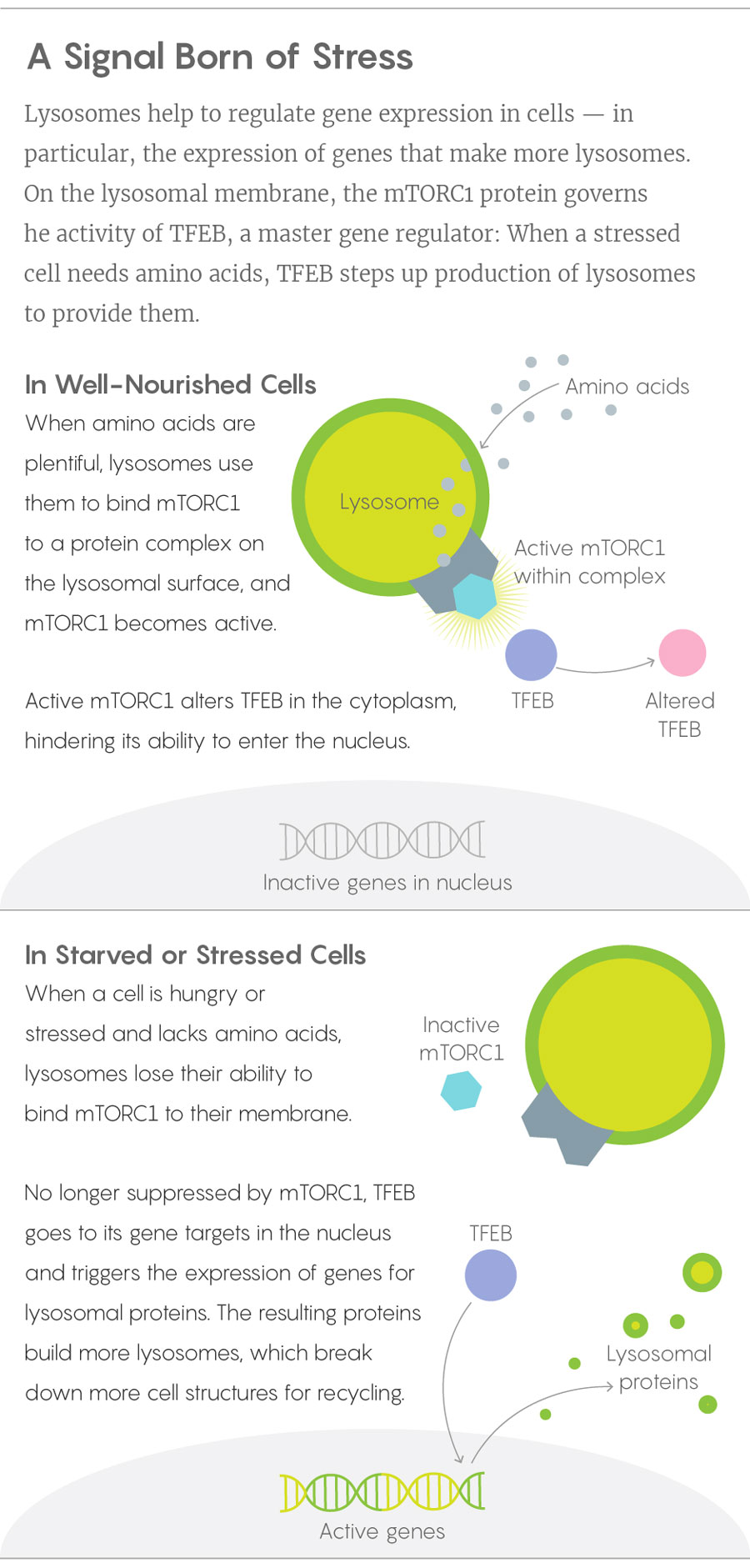
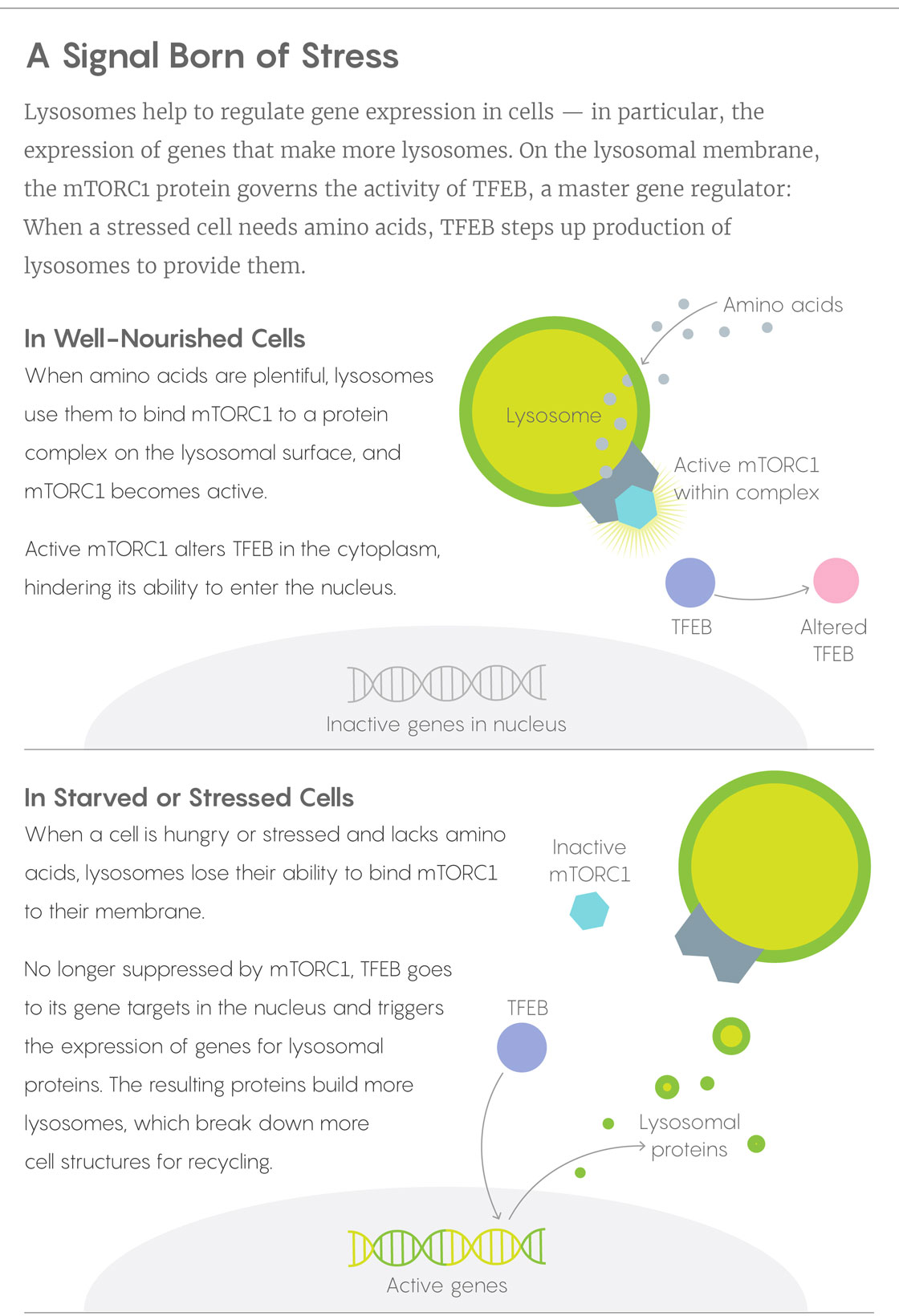
Lucy Reading-Ikkanda/Quanta Magazine
Starvation is not the only stressor that unleashes TFEB from lysosomes. Ballabio and his colleagues showed recently that TFEB can zip to the nucleus to help cells handle other stressful scenarios. According to Ballabio, several groups have shown that administering TFEB as a gene therapy in mice can help to alleviate symptoms of lysosomal storage diseases, diet-induced obesity and diabetes, and neurodegenerative conditions akin to Alzheimer’s disease and Parkinson’s disease.
Early in March, while chairing the Gordon Conference on lysosomes, Ballabio reflected over the phone to Quanta about how dramatically the field had changed. The field was once overwhelmingly about lysosomal storage diseases; now, he says, disease investigators mingle freely with those doing basic research. And the focus on deficiencies inside lysosomes had shifted to the lysosomal membrane and the ways in which it enlists TFEB, mTOR and roughly 200 other identified proteins in a conversation with the rest of the cell.
Cancer is one stubborn condition that might yield to a better understanding of lysosomes. Because cancer cells need plenty of nutrients to grow, “they have to reprogram or rewire their stomachs — their lysosomes — to take in and process a lot of food,” said Perera, a cancer biologist at the University of California, San Francisco.
She and Zoncu are jointly exploring the differences between lysosomes of malignant and normal cells. In addition, they are working to identify surface proteins on the lysosome that let nutrients escape to the cytoplasm for tumor cells to use. Such proteins could be exploited as portals for introducing toxins or drugs that cancer cells would collect in their lysosomes, to their detriment.
Another intriguing possibility comes from Ralph A. Nixon, a neuroscientist at New York University Langone Medical Center who also spoke at the lysosome conference. Experiments in his lab and others’ have linked failing lysosomes to cellular aging, reduced longevity and a range of neurodegenerative disorders. In 2015 Nixon and colleagues showed that certain gene mutations linked to Alzheimer’s disease disable a proton pump that maintains the pH of lysosomes. That change in acidity, which alters the balance of ions and metabolites that migrate out of lysosomes, can impair a cell’s metabolism.
Neurons may be uniquely vulnerable to this kind of low-level lysosomal disruption, Nixon says, and that could explain why lysosomal disorders so often have neurological consequences. If future drug interventions could correct problems with the lysosomal proton pump, or with other disruptions of lysosome function, it might be possible to stave off some of the neurodegenerative effects of Alzheimer’s disease or other conditions. Several drug compounds that modify lysosomal function — either by increasing rates of autophagy, or by raising rates of lysosome creation, or both — have shown promise against neurological conditions in mice, Nixon says.
Lysosomes may once have seemed like garbage bins that were “boring on the outside,” Nixon says, but they are increasingly appreciated as regulated signal platforms crucial to cellular health. And as perspectives on the lysosome change, views of the associated biology shift, too. Perera notes that cancer researchers have long wanted to know more about the signals that let malignant cells grow and multiply nonstop, and about how the cells co-opt nutrients. The new view of lysosomes, she says, reveals that these are “all different aspects of the same problem.”